第三节 基因突变致酶活性异常
由于基因突变导致酶活性降低或增高所引起的疾病称为遗传性酶病(hereditary enzymopathy)。遗传性酶病与分子病的区别在于后者引起机体功能障碍是蛋白质分子变异的直接后果;而前者则由于合成酶蛋白结构异常或调控系统突变后导致酶蛋白合成数量减少,通过酶的催化作用间接导致代谢紊乱所引起的机体功能障碍。基因突变导致酶的遗传变异可表现为酶活性降低、酶活性正常(同义突变或突变部位不影响酶活性中心)及酶活性增高。绝大多数遗传性酶病是由于酶活性降低引起的,仅少数表现为酶活性增高。
据估计,人类的酶有10 000种左右,但目前已明确的酶仅200多种,只占酶总数的2%左右。遗传方式以常染色体隐性遗传为多见,常染色体显性和X连锁隐性遗传者较少。杂合子所产生酶量往往等于正常纯合子和突变基因纯合子所含酶的半量,这种现象称为基因的剂量效应(gene dosage effect)。
一、酶活性降低引起的遗传性酶病
1.酶活性降低的原因基因突变引起酶活性降低的可能原因包括:①结构基因突变;其结果致使酶动力学特性改变(表现为酶与底物亲和力降低,与抑制物的亲和力增高)和酶的稳定性降低(表现为酶降解速率加快);②调节基因突变:酶合成速率减慢;③影响翻译后修饰和加工。
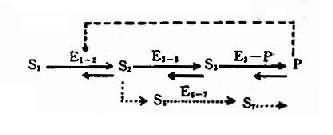
图表-21 典型代谢途径示意图
S1:底物;S2、S3:中间产物; P:终产物;E1-2、E2-3:不同代谢环节的酶;
E3-P:形成终产物的酶;E6-7:旁路代谢的酶;S6、S7:
旁路代谢产物;---旁路代谢途径;---反馈抑制
2,酶活性降低的发病机理 人类体内一般代射过程如图4-21所示:代谢底物(S1)进入细胞后,底物(S1)在一系列酶(E+1-2。E2-3,E3-P)催化下经过一系列中间产物(S2、S3),最后形成代谢终产物(P),同时代谢产物(P)可对一定的酶(如E1-2酶)起反馈抑制作用。另外,在正常代谢过程中还可能存在代谢旁路(S2→S6→S7……)。当控制酶蛋白合成的基因发生突变时,正常代谢受阻,可能通过不同发病环节引起不良后果。
(1)酶缺乏致代谢中间产物堆积和排出引起的疾病:当E2-3缺乏时,中间产物S2不能转变为S3,它在血和尿中的浓度增加,如果中间产物是无毒的,可由肾排出或通过其它方式降解,则不会严重危害人体。如尿黑酸尿平患者,因酪氨酸代谢中间产物(尿黑酸)堆积,可大量由尿排出,对人体无影响。但如果中间产物具有毒性,则将会引起症状。
半乳糖血症可作为实例,半乳糖血症(galactosemia)表现为婴儿哺乳后呕吐、腹泻,对乳类不能耐受,继而出现肝硬化、白内障、智力发育不全等症状。乳类含有乳糖,它经消化道乳糖酶分解产生葡萄糖及半乳糖,半乳糖通过一系列酶促反应产生葡萄糖而被组织利用(图4-22)。
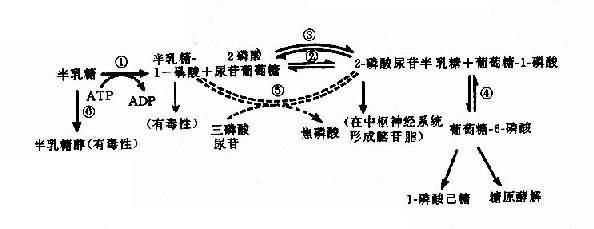
图4-22 半乳糖代谢途径
(虚线表示年长以后才发展起来的代偿途径)
①半乳糖激酶;②半乳糖-1-磷酸尿苷酰转移酶;③半乳糖尿苷-2-磷酸-4-异构酶;④磷酸葡萄糖变位酶;⑤-磷酸尿苷半乳糖(或葡萄糖)焦磷酸化酶;⑥醛糖还原酶
典型的半乳糖血症患者由于半乳糖-1-磷酸尿苷酰转移酶(galactose-1-phosphate uridy1 transferase,简称转移酶)缺乏(图4-22②),致使半乳糖-1-磷酸(Gal-1-P)及半乳糖积聚在血中,部分随尿排出。Gal-1-P在肝的积聚可引起肝功能损害,甚至肝硬化;在脑的积聚引起智力障碍;血中半乳糖升高可使葡萄糖释出减少,出现低血糖症。半乳糖在醛糖还原酶作用下产生半乳糖,能改变晶状的渗透压,使水分进入,影响晶状体代谢而致白内障。
患者都是隐性纯合子(gg),杂合子表型正常,转移酶活性约为50%,活性低于10%可出现典型症状。
另一类半乳糖血症为半乳糖激酶(galactokinase)缺乏所引起,症状较轻,主要表现为青年型白内障,血中半乳糖增高,但无肝及脑损害。
转移酶基因(GALL)定位于9p13,半乳糖激酶基因(GALK)定位于17q21-q22。两病均为常染色体隐性遗传。
(2)酶缺乏致代谢底物堆积引起的疾病:当一系列生化反应可逆时,一处的阻断常导致代谢底物(S1)贮积。贮积的物质如果溶解度高,则在血和尿中浓度增高;若溶解度低时,则在组织中贮积引起疾病。
糖原贮积症(glycogen storage liseaee,GSD)是一组由糖原合成和降解酶缺陷引起的疾病,至少有12种类型。糖原贮积症主要累及肝或肌肉,但有的也可伴有心、肾和神经系统的损害。不同类型之间其严重性和预后都不全相同。例如,von Girke病(Ⅰ型)症状非常严重,而Hers病(Ⅵ型)较轻。此类疾病发病机理可用von Gierke病为例说明。本病是由于肝内葡萄糖6-磷酸酶(glucose-6-phosphatase,G6Pase)缺乏引起。肝糖原在一系列酶的作用下生成葡萄糖,这个反应的各个步骤都是可逆的,其主要步骤如下:
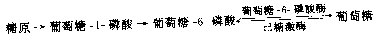
患者由于G6Pase缺乏,所以G6P不能转变为葡萄糖供组织利用,通过可逆反应而合成过多的肝糖原,引起患儿肝肿大。当不进食时极易发生低血糖。由于动用脂肪可以出现酮血症。G6P通过无氧酵解,生成大量乳酸,导致酸中毒。所以患者的肝大伴低血糖,发育不良,消瘦,身体矮小,常有出血倾向。肝活检见糖原含量增加。
本病呈染色体隐性遗传。患者G6P酶完全缺乏,多数患者父母表型正常,但G6P酶活性为中间值。同胞中可出现患者。
(3)酶缺乏致代谢终产物缺乏引起的疾病:这是一类由E2-3酶缺乏,致所有产物(包括终产物P)缺乏引起的疾病.白化病就是实例.白化病(albinism)分全身型及局部型.全身型常见,患者皮肤呈白色,毛发银白或淡黄色,虹膜及瞳孔呈淡红色,视网膜无色素,羞明,眼球震颤等,本病发病率约1/10000-1/20 000,呈常染色体隐性遗传.
正常人黑色素由黑素细胞合成,这些细胞中有特殊的细胞器-黑素小体(melanosome),其中有含铜的酚氧化酶,即酪氨酸酶(tyrosinase),它可将酪氨酸转变成黑色素(图4-23).白化病人有黑素细胞,但酪氨酸酶缺乏,使黑色素不能形成,病人因缺黑色素而白化.白化病存在遗传异质性.已知白化病至少有7种不同类型.酪氨酸酶基因(TYR)定位于11q14-q22.
属于此类的例子尚有遗传性甲状腺肿,它是由于偶联酶、脱碘酶或碘过氧化酶缺乏导致甲状腺素生成减少引起的,患者身材矮小,智力低下,面貌丑陋.
(4)酶缺乏致旁路产物增多引起的疾病:当酶的缺乏导致主要代谢途径受阻断时,过量的前体物S2通过另一旁路代谢引起某些副产物的堆积.如果增多的旁路产物是无毒的,则可排出体外不致危害人体,但如果旁路产物或其分解产物有毒,则可危害机体引起疾病.
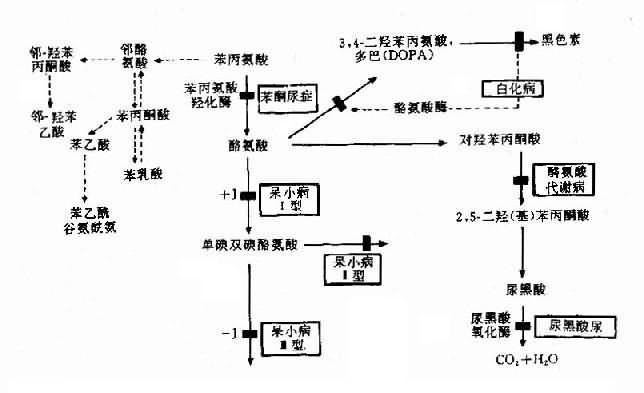
图4-23 苯现氨酸代谢及有关的遗传性酶病的发病机理
苯酮尿症(phenylketouria,PKU)可作为实例。本病以智能发育不全为主要特征,呈常染色体隐性遗传,群体发病率约1/16000,由苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)遗传性缺乏引起。现已知苯丙氨酸羟化酶基因定位于12q24.1,此基因全长约90kb,含13个外显子,在中国人中已发现10余种点突变,这是造成酶活性缺乏的原因。
典型PKU患儿出生时,外貌正常,约至3-4个月时,渐出现智能发育不全,患儿步伐小,姿似猿猴,肌张力增高,易激动,甚至惊厥,90%以上患者毛发发黄,肤白,甚至虹膜呈黄色(白种人呈蓝色)。此外,患儿尿和汗有一种特殊的腐臭。
正常人苯丙氨酸在体内主要通过PAH的作用转变成为酪氨酸,继而生成黑色素(图4-23)。PAH主要存在于肝中,它的作用需要辅因子四氢生物蝶啶(BH4),如果PAH缺乏(活性<1/10)可阻断苯丙氨酸转化为酪氨酸,即产生典型的苯酮尿症。酶活性部分缺乏,可导致轻度高苯丙氨酸血症(hyperphenylalaninemia)。
典型PKU患者,由于肝内PAH几乎完全缺如,致血清苯丙氨酸增高(正常人1-3mg%,患者可达50-100mg%)。过多的苯丙氨酸通过转氨酶作用生成苯丙酮酸,再经氧化.脱羧产生苯乳酸.苯乙酸等异常产物,由尿和汗液排出,致患儿的尿和汗呈特殊的腐臭。可能由于旁路产物抑制了脑组织内L-谷氨酸羧酶,使谷氨酸脱羧基生成γ-氨基丁酸减少,而后者对脑细胞的发育及功能起重要作用。也有人认为苯现氨酸和旁路产物可影响5-羟色胺的生成,而影响脑功能。由于正常产物酪氨酸是黑色素前体,所以酪酸不足加之旁路产物可以抑制酷氨酸酶,使患者皮肤毛发及眼血管膜色素减少,因而色泽都较浅。
(5)酶缺乏致反馈抑制减弱引起的疾病:有些代谢过程中,某些代谢产物对整个反应过程具有反馈调节作用。因此,某种酶的遗传性缺陷,使该代谢产物减少,致反馈调节功能失调。自毁容貌综合征可作为实例。处毁容貌综合征亦称Lesch-Nyhan综合征(Lesch-Nyhan syndrome,LNS),是由于遗传性次黄嘌呤鸟嘌呤磷酸核糖转移酶(hypoxanthine-guanine-phosphoribosyl transferase,HGPRT)缺乏所引起。本病的特征是智力发育不全、舞蹈样动作和强迫性自残行为,并伴有高尿酸血症、尿酸尿、血尿、尿道结石和痛风。患者可活至20余岁,多死于感染和肾功能衰竭。
LNS呈X连锁隐性遗传,患者均为男性(半合子)。现知HGPRT定位于Xp26-q27.2,其发病率约为1/380000。
正常时,HGPRT的功能是将次黄嘌呤转变为次黄苷酸(((即IMP,肌苷酸),将鸟嘌呤转为鸟苷酸(GMP),鸟苷酸及腺苷酸可以反馈抑制磷酸核糖焦磷酸(PRRR)合成1-氨基-5-磷酸核糖,从而控制IMP的自发合成速度(图4-24)。这一过程,HGPRT的催化起到反馈抑制作用。当HGPRT缺乏时,这一反馈抑制作用减弱或消失,IMP和GMP生成量大为减少,嘌呤合成速度就大大增快,致使患者体液内的尿酸浓度增高,出现高尿酸血症和尿酸尿。经典型患者无HGPRT活性。酶仅部分缺乏时,患者往往出现痛风而无LNS症状。
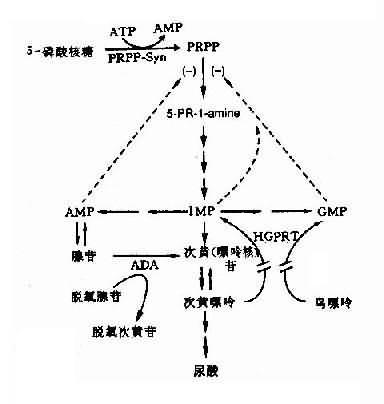
图4-24 HGPRT缺乏导致Lesch-Nyhan综合征
5-PR-1-amine:-1氨基-5-磷酸核糖;
PRPP-Sym:磷酸核糖焦磷酸全盛酶;
虚线:反馈抑制
(6)维生素依赖性遗传病:这是由于基因突变改变了某种酶蛋白,使之与辅酶的相互作用受到损害引起该酶活性降低。这类辅酶多数为维生素,故称为维生素反应性遗传病(vitam-in-responsive hereditary disorders)。例如,在体内,吡多醇-5-磷酸(PLP)是维生素B6的活性形式,它是多种酶蛋白的一种辅酶,因此,某些酶蛋白基因的突变能引起多种遗传性代谢病(图4-25)。例如谷氨酸脱羧酶基因的突变致使酶蛋白和PLP的相互作用受损,将导致γ-氨基丁酸缺乏和痉挛。胱硫醚合成酶基因突变使酶蛋白与PLP的结合部位发生改变而导致胱硫醚尿症。胱硫醚合成酶的缺乏可导致同型胱氨酸尿症。
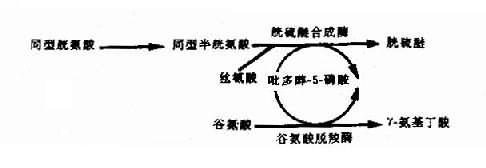
图4-25 吡多醇-5-磷酸(PLP)作为多种酶的辅酶形式示意图
(7)多种酶缺陷引起的疾病:有些遗传性代谢缺陷,在某一患者不只一种酶缺陷。例如先天性蔗糖不耐受症(congenital sucrose intolerance),患者其异麦芽糖酶和蔗糖酶都缺乏,而枫糖尿症(maple syrup urine disease)病人体内则同时缺乏缬氨酸脱羧酶.亮氨酸脱羧酥酶和异亮氨酸脱羧酶。这种现象可作如下解释:①有缺陷的几种酶均有一条共用的多肽链,当编码这条多肽链的结构基因发生突变时,就会使凡含有这条共同多肽链的各种酶蛋白结构改变而失活;②由于一个酶,致使由这种催化生成的代谢物缺乏,因此由它诱导的各种酶也相应缺乏;③由于一个调控基因发生突变,关闭了邻近几个结构基因,使这些结构基因控制的酶不能产生。
二.酶活性增高引起的遗传性酶病
基因突变可使个别酶活性增高,其代谢产物增多从而可引起疾病。例如有一类痛风(gout)是由于患者体内的磷酸核糖焦磷酸合成酶(phosphoribodsyl pyrophosphate synthetase,PRPP-syn)结构变异,酶活性可升高3倍,致使磷酸核糖焦磷酸(PRPP)的生成大大加快(图4-24),结果由PRPP分解的终产物尿酸在体内大量增加,并从尿中排出,由于尿酸的溶解度低,过多的尿酸盐在结缔组织中沉积,出现痛风性关节炎,久后造成关节变形,大量尿酸在尿中沉积,可形成尿路结石。
目前已知PRPP-syn活性增高有三种情况:①酶分子合成正常,但每分子酶的活性增高2-3倍;②PRPP-syn与底物5’-磷酸核酮的亲和力增高;③酶与抑制物的亲和力降低。本病呈常染色体显性遗传。PRPP-syn酶基因定位于Xq22-q26.
酶活性过高导致产物过多引起的一类疾病称为生产过剩病(over-production disease)。